
Thanks to Caitlin for organising this year’s Secret Santa/White Elephant.
Thanks to Caitlin for organising this year’s Secret Santa/White Elephant.
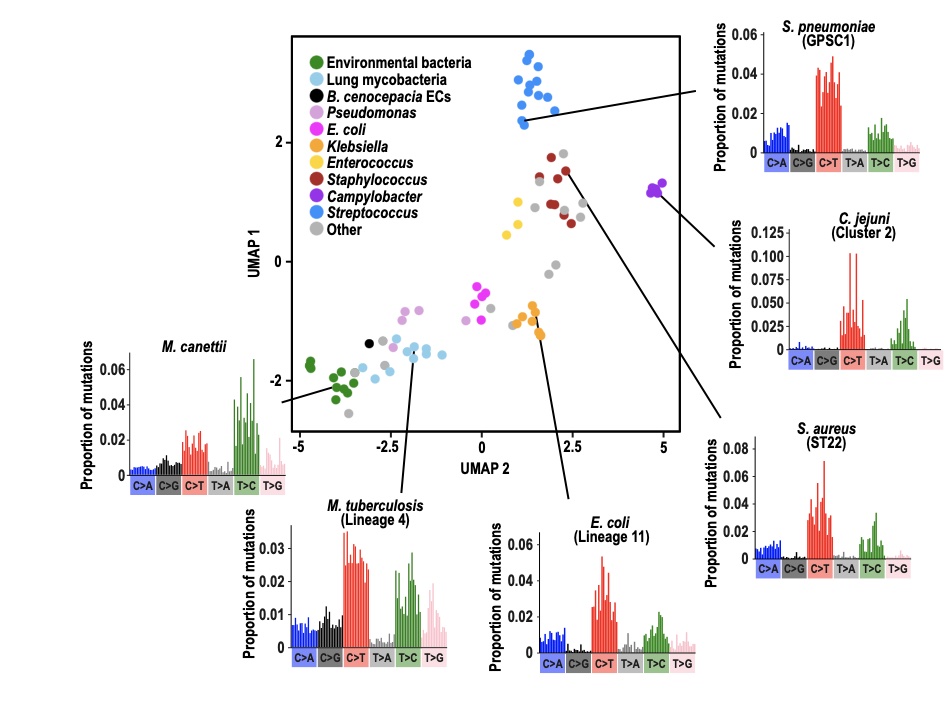
Led by Chris Ruis and a collaboration with Andres Floto in the Department of Medicine, our new paper entitled Mutational spectra analysis reveals bacterial niche and transmission dynamics is now on bioRxiv.
Mutational spectra (the patterns of base mutations and their surrounding nucleotide contexts) have been widely applied within the cancer field to infer the mutagenic drivers of cancers. This work has shown that individual mutagens drive specific mutational patterns. We reasoned that bacteria that live in different niches will be exposed to different sets of mutagens that will cause different mutational patterns. Therefore by charting the mutations acquired during evolution, we may be able to infer where a bacteria has lived and therefore the source(s) of infections. We tested this by calculating mutational spectra of 84 phylogenetic clades across 31 different bacterial species using a specifically developed pipeline, MutTui. This showed that mutational spectra are highly diverse across bacteria, both in base mutations and their contexts. However, we could observe groups of similar spectra that typically belonged to bacteria that are closely related and/or live in the same niche.
We examined the ability of mutational spectra to predict niche across several levels of comparison. We first showed that the mutational spectra of lung and environmental clades of Mycobacteria and Burkholderia cluster by niche. This is likely driven by elevated C>A mutations in the lung and higher T>C mutations in the environment. A decomposition analysis on the niche-specific mutations suggested that higher C>A in the lung is likely driven by tobacco smoke and reactive oxygen species, while elevated T>C is driven by environmental alkylating agents and nitro-PAHs. We next examined multiple niches within a single host and found that enteric and invasive lineages of Salmonella exhibit distinct spectra. Finally, we examined spectra of skin bacteria and identified a UV light-associated signature in the pan-skin bacterium Cutibacterium acnes that is not present in Staphylococcus epidermidis, which inhabits moist skin sites and is therefore hidden from the external environment.
Together, these results show that mutational spectra can predict general and specific bacterial niches and therefore reveal pathogen infection sites and transmission routes.

Last week members of the lab went punting along the Cam.

Led by Noémie Lefrancq and Valérie Bouchez, and a collaboration between the Department of Genetics, the Vet School, Institut Pasteur Paris, and the EuPertStrain consortium, a new paper entitled Global spatial dynamics and vaccine-induced fitness changes of Bordetella pertussis is out now in Science Translational Medicine.
The study was supervised by Sylvain Brisse from Institut Pasteur Paris, and Henrik Salje from Department of Genetics at the University of Cambridge.
Bordetella pertussis (Bp), which causes whooping cough, infects >24 million individuals annually despite widespread vaccination. Asymptomatic carriage and multiple circulating lineages hide the underlying dynamics of Bp from surveillance systems. Therefore, the extent of spread across spatial scales remains a mystery, as does the role of vaccines in driving changes in strain fitness. In this study we use models informed by pathogen sequences to help answer these key questions.
We analysed 3,344 genomes from 23 countries, covering an 85-year period, thanks to the establishment of the EuPertStrain consortium of national reference laboratories and publicly available data GenBank. We found substantial diversity within communities, consistent with widespread subclinical transmission, and a strong association between the number of transmission chains circulating within subnational regions and the host population size. Bp strains are able to spread both nationally and internationally in just a few years, e.g. it takes only 5-10 years for individual lineages to be homogeneously distributed throughout Europe or the United States. Next, we developed an analytical model that estimates the relative fitness of different genotypes across countries. Heterogeneity in vaccine introductions provides a natural experiment to characterize fitness changes following switches in the local vaccine type being used. Our framework recovered the substantially different genotype dynamics that have been observed across countries, enabling us to estimate the relative fitness of the different genotypes, by vaccination era. We found that increased fitness of pertactin-deficient strains following implementation of acellular vaccines, but reduced fitness otherwise, can explain the long-term genotype dynamics.
This work provides a first detailed view of the strength of spatial structure and rate of geographic spread, highlighting how Bp spread is a globally interconnected issue. It further provides a quantitative description of strains fitness and highlights the key role of vaccine policy decision-making in driving ecological change.
Our new preprint “Mycobacterium avium complex (MAC) genomics and transmission in a London hospital” (https://www.medrxiv.org/content/10.1101/2022.01.07.22268791v1) is now out.